
Performance Metric Report 2017
Develop improved open access platforms for computational analysis of large genomic datasets.

Read on for a summary of KBase FY17 quarterly reports, included here:
Q1: Report on the latest capabilities for annotating and assembling genome-based metabolic models of microbial metabolism [PDF]
Q2: Report on the new capabilities to perform reproducible genomics analyses on large datasets and share the results with other researchers [PDF]
Q3: Describe the latest developments for working with and analyzing plant genomes [PDF]
Q4: Report on new capabilities and approaches for analyzing metagenomics datasets [PDF]
1. Introduction
The DOE Systems Biology Knowledgebase (KBase, www.kbase.us) is an open-access software and data platform designed to integrate large, heterogeneous data into predictive models of biological function ranging from a single gene to entire organisms and their ecologies. In a single integrated environment, KBase enables users to create, execute, collaborate on, and share sophisticated, reproducible analyses of their biological data in the context of public data and data other users have privately shared with them [1].
KBase’s features include: (i) comprehensive support for data provenance and analysis reproducibility; (ii) a flexible system for sharing data and workflows; (iii) an internally maintained database of genomes and biochemistry; (iv) a point-and-click interface (the Narrative Interface) that enables users to build, store, run, and share complex scientific analyses of fully integrated data; (v) built-in support for the use of custom code interleaved with point-and-click apps; and (vi) a software development kit (SDK) that allows external developers to add apps to KBase. KBase has a growing suite of scientific tools that enable users to build and share sophisticated workflows-–for example, one can predict species interactions from metagenomic data by assembling raw reads, binning assembled contigs by species, annotating genomes, aligning RNA-seq reads, and reconstructing and analyzing individual and community metabolic models. That is one example of a workflow supported by KBase; there are numerous branch points, alternative pipelines, alternative entry points, and internal curation loops that provide for a wide range of scientific analyses, some of which are not available elsewhere (e.g., merging individual metabolic models into community models and using these to predict interspecies interactions).
The support for collaboration and reproducibility in KBase is centered around the Narrative Interface, which lies at the core of the KBase user experience. Built on the Jupyter Notebook [2] framework, the Narrative Interface (Figure 1) supports both point-and-click and scripting access to system functionality in a “notebook” environment, enabling computational sophisticates and experimentalists to collaborate within the same platform and share their datasets and workflows. Each Narrative in KBase is an interactive, reproducible document that allows users not only to see what a researcher did, but to quickly and easily replicate and even extend their analyses.
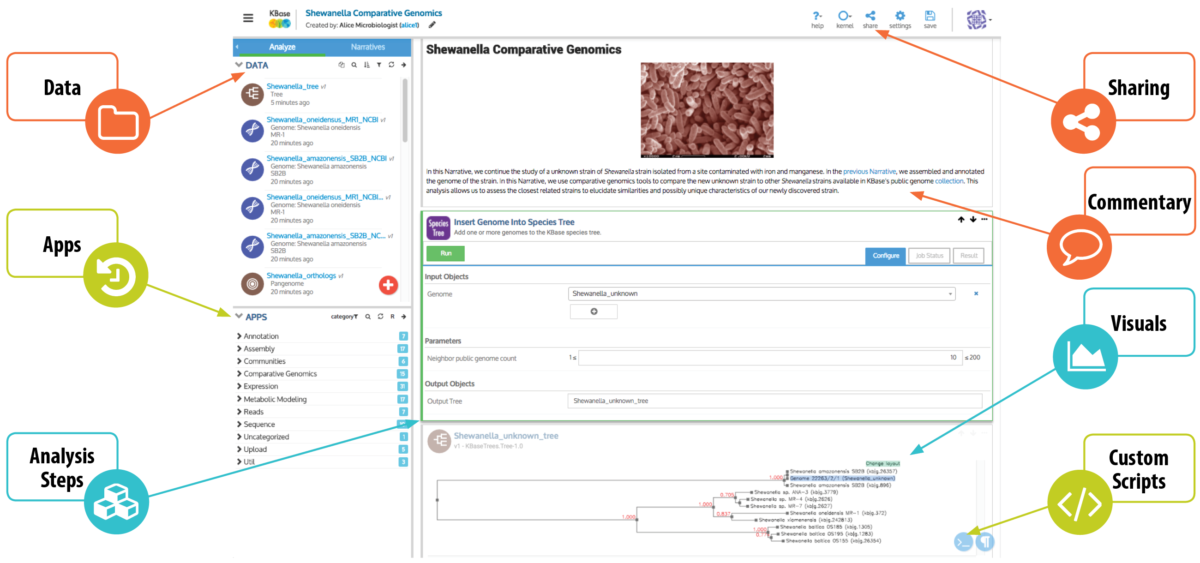
Figure 1. KBase Narrative. A Narrative is a shareable, reproducible computational experiment that can include data, analysis steps, results, visualizations and commentary.
The web-accessible KBase system (https://narrative.kbase.us) runs on DOE computing infrastructure and is open and freely available for anyone to use. KBase’s code, available at github.com/kbase, is open source and distributed under the MIT License. KBase adheres to the FAIR (Findable, Accessible, Interoperable, Re-usable) data principles endorsed by many funding agencies and scientific organizations [3].
2. Capabilities for computational analysis of large genomic datasets
KBase has a growing and extensible set of interconnected applications (apps) for data upload and download, reads processing, contig assembly, genome annotation, metabolic model reconstruction, flux balance analysis, expression analysis, metagenomics and comparative genomics (Figure 2). New capabilities in KBase for bulk upload and execution make it possible to upload large datasets and quickly run them through sophisticated analysis workflows.
KBase facilitates large-scale analysis by providing ways to upload large individual data files and large collections of data, as well as enabling bulk processing (as described in the Q2 report). Currently, KBase supports bulk upload of sequencing reads (Paired End, Single End, SRA) and genomes (GenBank files) through its Narrative “staging” interface; support for additional data types is in progress.
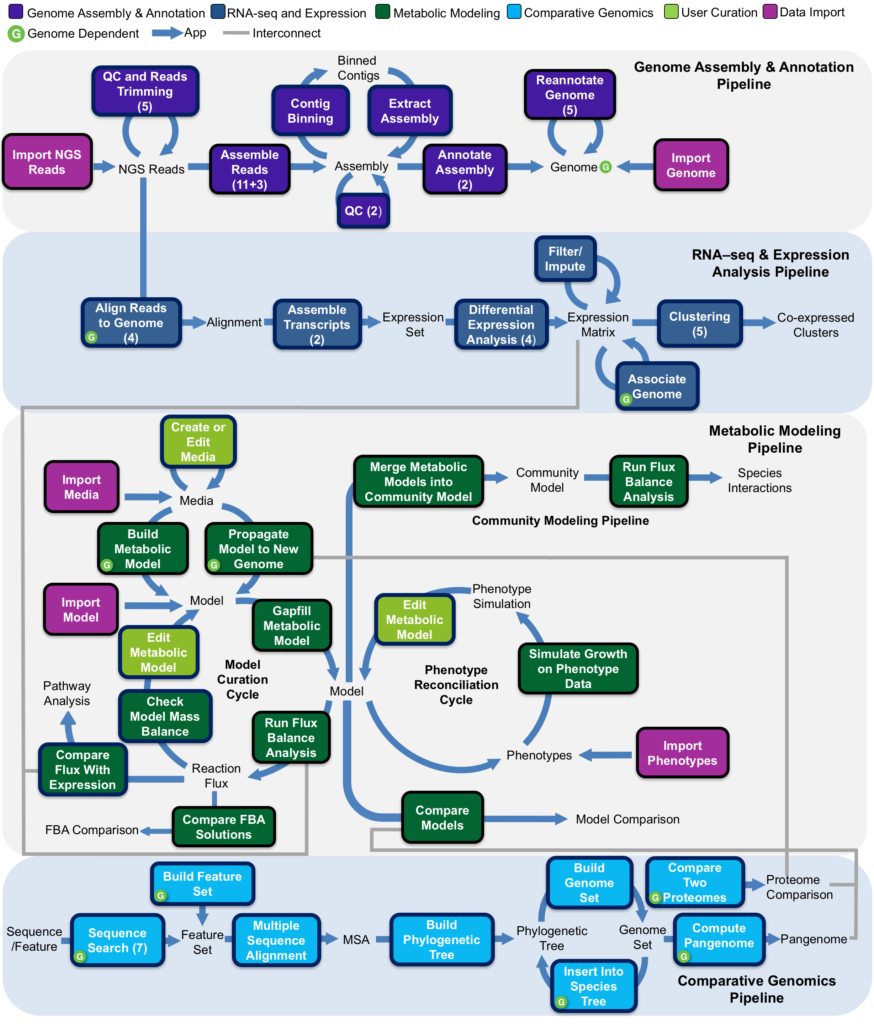
Figure 2. Some of the workflows and data types currently available in KBase.
One of the advantages of building the Narrative Interface on the Jupyter Notebook framework is the support for “code cells”, which enable any user to write, execute, and share custom code from within KBase’s graphical user interface. Because code cells support programming paradigms such as loops, they can be applied to run apps in bulk, which can be very useful when processing a large dataset. KBase also offers some apps that run specific analyses in bulk, such as Annotate Multiple Microbial Genomes (see section 2.3).
KBase has 14 apps for assembling Next-Generation Sequencing (NGS) short reads and generating annotated genomes from these assemblies. The starting point for assembly in KBase is a set of single- or paired-end reads generated from a variety of sequencing technologies, including Illumina, PacBio CLR, PacBio CSS, IonTorrent, and Oxford Nanopore (see https://docs.kbase.us/workflows/assembly-annotation). A new app (in beta) wraps HipMer [4], a fast “extreme-scale” assembler suited for large genomes such as those of plants. Before assembling reads, it may be beneficial to preprocess them to remove adapters, barcode sequences and low-quality reads; KBase offers several apps for this purpose, including one in development that uses the JGI’s reads data preprocessing pipeline (BBtools [5]).
KBase recently added apps that run several of the most commonly used metagenome assemblers (MEGAHIT [6], metaSPAdes [7], and IDBA [8]). These metagenome assemblers produce “Assembly” objects that are groups of contigs that come from the mixture of species present in the samples. KBase provides the ability to compare the distributions of the contigs produced by the assemblers with the QUAST [9] tool.
KBase supports microbial genome annotation via the RAST (Rapid Annotations using Subsystems Technology) pipeline [10], which assigns functions from the SEED Subsystems Ontology [11] to genes, and also the Prokka prokaryote annotation tool [12]. The Annotate Multiple Microbial Genomes app makes it easy to run the RAST pipeline on a number of genomes at once. The Annotate Plant Transcripts with Metabolic Functions app assigns predicted functions to plant transcript sequences. The annotations generated by these apps can be explored in a tabular genome viewer or exported in GenBank format.
KBase has an extensive suite of apps and data that support the reconstruction, optimization and analysis of metabolic models for microbes, plants and their communities (see https://docs.kbase.us/workflows/metabolic-models); these were covered in detail in our previous KBase highlights and are summarized briefly here. In KBase, genome-scale metabolic models are primarily reconstructed from functional annotations produced by the KBase annotation apps, which map metabolic genes onto biochemical reactions. This information is integrated with data about reaction stoichiometry, subcellular localization, biomass composition, directionality of reactions, and other constraints into a detailed stoichiometric model of metabolism.
Metabolic modeling in KBase begins with the Build Metabolic Model app, which uses the functional annotations to generate a draft metabolic model [13]. Draft metabolic models usually have missing reactions (gaps) due to incomplete or incorrect functional annotations making them unable to produce biomass when simulating growth conditions in which the organism should be viable. The Gapfill Metabolic Model app in KBase overcomes this problem by identifying the minimum number of new reactions that must be added to the model, or existing reactions that must be made reversible, to enable the production of biomass [14].
After building and gapfilling a metabolic model, researchers typically run Flux Balance Analysis (FBA) to predict reaction fluxes and optimal growth or metabolite production yields. The Run Flux Balance Analysis app in KBase applies the FBA approach to predict the flow of metabolites through the metabolic network of an organism by optimizing a selected cellular objective function, typically biomass maximization. This app requires the user to specify a media formulation in which the growth will be simulated. KBase currently maintains more than 500 commonly used media conditions, and also allows users to upload their own custom media formulations. The Run Flux Balance Analysis app also optionally accepts a gene expression profile as input.
After running the Run Flux Balance Analysis app, two or more models can be compared with the Compare Models app. Alternatively, the Compare FBA Solutions app can be used to compare flux profiles predicted by FBA to understand how an organism’s behavior changes from one condition to the next, or how the behavior of two different organisms differs within a single condition. Another useful modeling app is Simulate Growth on Phenotype Data, which takes as input a list of growth phenotypes for simulation. In this case, we define a growth phenotype as an indication of growth rate for an organism on a particular growth condition (media formulation) that may be affected by a particular set of gene knockouts. KBase also now offers an embedded metabolic model editor that allows users to interactively add, remove, or change compounds, reactions, or biomass.
The metabolic modeling tools in KBase can be used to model eukaryotes such as plants, as well as microbes and other prokaryotes. KBase also can construct a merged metabolic model to study interactions between different tissues and organs within a single species, or cross-species interactions between multiple organisms (which may include microbes and plants). This approach has been applied extensively to predict potential interactions in a variety of systems, some of which are described in Section 3.
RNA-seq analysis is useful for assessing differential gene expression. It can shed light on questions such as which genes are over- or underexpressed at various stages of development, which genes are expressed differently in a disease state compared with normal cells, or how changing environmental conditions such as temperature changes, drought, or differences in soil chemistry can affect the production of plant biomass. The overarching goal of the RNA-seq pipeline in KBase is to create differential expression estimates and use these to inform metabolic models and to perform functional analysis of genes with similar expression patterns.
RNA-seq analysis typically consists of (i) aligning reads to the reference genome; (ii) assembling transcripts and quantifying expression; and (iii) differential analysis of the gene expression. KBase provides a set of apps that allow users to run the tools from the popular Tuxedo RNA-seq suites [15,16] to generate the normalized full and differential expression matrix of the reads obtained from Illumina sequencing platforms using the reference prokaryotic and eukaryotic genome. The RNA-seq apps in KBase can be combined into multiple workflows, allowing users to select their choice of reads aligner and assembler for the differential gene expression analysis (see https://docs.kbase.us/workflows/rnaseq).
Comparative genomics is a growing area of functionality in KBase. KBase has apps in active development to enable gene family analysis within and across species, including BLAST-based homology search, multiple sequence alignment, and construction of phylogenetic trees. Phylogenetic reconstruction in KBase works in three steps: 1) generate a feature set (a list of genome references, used by the next steps in the pipeline); 2) multiple sequence alignment; and 3) build phylogenetic tree. There are apps in development for generating feature sets via a BLAST search against genomes or by searching the gene annotation/identifiers. A feature set can be used as input for multiple sequence alignment, and in turn, a multiple sequence alignment can be used to build a phylogenetic tree (FASTTREE-2 Phylogenetic Tree from MSA [17]; see Figure 3).
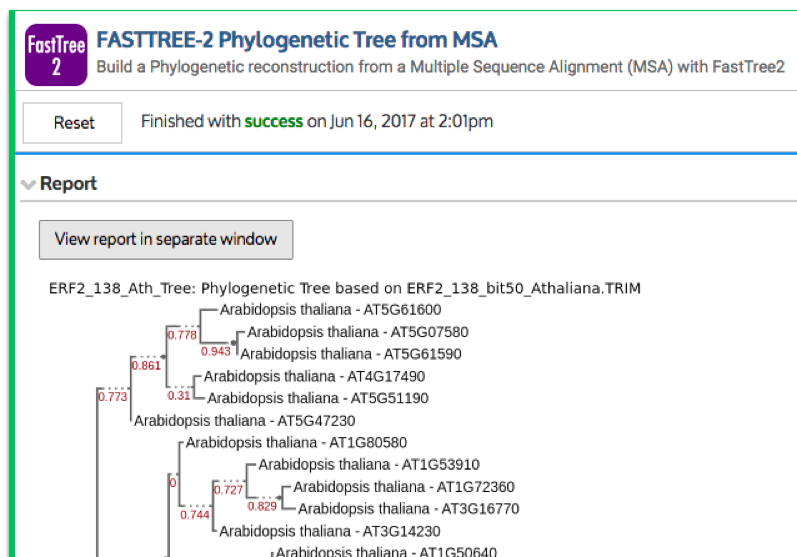
Figure 3: Part of the phylogenetic tree for the ERF protein family members in A. thaliana as constructed by the “FASTTREE-2 Phylogenetic Tree from MSA” App.
The Narrative entitled “Discovery and characterization of ERF protein family members in Arabidopsis thaliana” (https://narrative.kbase.us/narrative/22292) exploits some of the comparative genomics functionality in KBase to demonstrate genome-wide characterization of a particular gene family–a workflow that is widely used by plant biologists. The ERF family of genes encodes transcription factors that are involved in various developmental and physiological processes in plants. In this Narrative, the ERF gene family members in the model plant A. thaliana are analyzed and characterized. This approach is based on mining homologs of the tobacco AP2 domain containing the ERF2 protein in A. thaliana using BLASTP [18] and HMMER [19] searches. Altogether, 138 ERF protein family members were found in A. thaliana and a phylogenetic tree (Figure 3) was built based on this gene family.
A new suite of tools released in summer 2017 has streamlined the process of performing metagenomic analyses in KBase. A user can predict species interactions from metagenomic data by assembling raw reads, binning assembled contigs by species, annotating genomes, aligning RNA-seq reads, and reconstructing and analyzing individual and community metabolic models. Users have applied these tools to study: (i) interactions between plants and microbes in soil; (ii) why some microbes form stable communities; (iii) how a microbial community cooperates to produce a specific product; and (iv) how a community of heterotrophic species can feed on byproducts from an autotroph to grow autotrophically.
This new metagenome analysis pipeline, which is described in detail in the Q4 report, begins with the upload of large reads files into the user’s Narrative. Next, the user can apply one of three new apps for metagenome assembly (MEGAHIT, metaSPAdes, and IDBA). The assembled contigs can then be binned using MaxBin [20] and new genomes can be extracted from the bins. These extracted genomes can be analyzed for completeness using the genome quality analysis app (CheckM [21]). After metagenomic reads are assembled and binned into standard genomes, these can be piped into a wide range of downstream analysis apps in KBase, including genome annotation, genome comparison, metabolic modeling, and RNA-seq alignment. Some of these downstream analysis pipelines have recently been updated to improve support for metagenome analysis. A scientific use case showing a metagenomic analysis workflow in KBase is described in section 3.4.
3. Science use cases
KBase is being used by a growing number of scientific groups, with citations in over 30 peer-reviewed publications to date covering a range of topics including comparative genomics of plants, prediction of microbiome interactions, and deep metabolic modeling of environmental and engineered microbes. Several of these studies have publicly shared KBase Narratives associated with them. These show how the KBase platform was applied to perform the described work as well as serving a means of releasing the data produced in the published analysis. Here we will review a selection of these research Narratives that demonstrate many (but not all) of the workflows (see Figure 2) and capabilities available in the KBase platform.
Many published Narratives can be found on the KBase Research website (https://www.kbase.us/research); these can be viewed, copied, and re-run, possibly with different parameters or new datasets. By enabling reproducible scientific analysis on large datasets and facilitating collaboration, KBase is helping to accelerate the pace of systems biology research.
With its multiple apps for microbial genome assembly and annotation, KBase is an ideal platform for the analysis of new isolate genomes. This is demonstrated in a recent publication [22], in which groundwater samples were collected from multiple wells, where researchers identified a Janthinobacterium isolate that produces violacein. Violacein is a naturally-occurring bis-indole pigment with antibiotic (antibacterial, antifungal, and anti-tumor) properties. The team loaded their isolate reads into KBase, performing QC and assembly. They next applied both the Prokka and RAST annotation tools to the assembled genome. They also ran OrthoMCL [23] to compare the two alternative annotations side by side. By combining both sets of annotations, the researchers were able to identify and confirm the presence of all violacein genes in their isolate of interest. The analysis is captured in the linked Narrative (https://narrative.kbase.us/narrative/21546), including the researchers’ thought process at each step. This study demonstrates the value of having multiple apps for some of the key steps of the isolate analysis pipeline. During genome assembly and genome annotation, the researchers were able to apply multiple algorithms, evaluating and comparing the results and selecting the best-performing algorithm for each step.
KBase was applied to build core metabolic models (CMMs) representing central metabolism of bacteria for over 8000 genomes that span the prokaryotic tree of life [24]. The authors used CMMs to determine: (i) accurate ATP yields based on different growth/environmental conditions (ii) ETC variations and respiration types (iii) ability to produce fermentation products (iv) presence and absence of classical biochemical pathways in central metabolism and (v) ability to produce key metabolic pathway intermediates in central metabolism which are precursors of essential biomass components of the cell. KBase’s capacity for large-scale analysis enabled the researchers to quickly generate the 8000 CMMs, a task that would have been difficult or impossible without KBase. The complete CMM analysis workflow, using E. coli as an example to demonstrate bulk metabolic model construction using code cells, is captured in a public Narrative (https://narrative.kbase.us/narrative/20186). The models can be downloaded in bulk from that Narrative using the Bulk Download Modeling Objects app. This example demonstrate how KBase has been applied to run high-throughput analyses on thousands of genomes. Other high-throughput studies have also been performed in KBase, including the generation of draft models for 773 human gut microbes [25].
In a cross-kingdom community modeling study [26], researchers explored a mutually beneficial metabolic partnership between a moss and a bacterium, using KBase data and tools to build a merged community metabolic model in which a nitrogen-fixing diazotrophic microbe (Anabena) helps a plant (Sphagnum, or peat moss) to grow. All the analysis steps were performed in KBase (https://narrative.kbase.us/narrative/9667), culminating with merging the two models into a community model which exhibits nitrogen fixation and exchange, showing that the plant portion of the model consumes the nitrogen fixed by the microbial portion of the model, and predicting that the Sphagnum will grow more when utilizing nitrogen fixed by the microbe than when fixing nitrates on its own. This experiment utilizes the plant reference genomes in KBase, as well as the plant model reconstruction pipeline. It also shows how models of any type (for example, microbial and plant models) can be merged together in KBase to form a community model.
In a recent published study [27], researchers applied KBase annotation and metabolic modeling pipelines to analyze a 13-species electrosynthetic community that captures electrons from a cathode and fixes carbon dioxide. Metabolic models of the predominant community members belonging to Acetobacterium, Sulfurospirillum, and Desulfovibrio revealed that Acetobacterium is the primary carbon fixer for the community, excreting large amounts of acetate which serves as the main carbon source for the rest of the community.
To perform this analysis, the researchers selected the three most abundant members from this 13-species community based on the relative abundance data (Acetobacterium, Sulfurospirillum, and Desulfovibrio) and imported their genomes into KBase, followed by metabolic model reconstruction, gapfilling, and metabolic flux analysis of each model. The researchers then used the Compare Flux with Expression app to assess the agreement of the model predictions with reactions based on differentially expressed genes. The analysis workflow for this study, including trophic interactions predicted between these three species based on metabolic model analyses, can be found in https://narrative.kbase.us/narrative/15248.
When this electrosynthesis analysis was originally performed, KBase lacked any tools for metagenome assembly and binning, so those steps had to be performed outside of the system. Now that those tools have been added to KBase, it became possible to perform the entire experiment in a single integrated platform. The original raw metagenomics sequencing data was uploaded into KBase, followed by de novo assembly of the community, contig binning, genome quality assessment, iterative annotation of each of the assemblies (bins), and species tree analysis to identify the closest taxonomic lineage. Based on the tree analysis data, the three newly binned genomes were found to closely resemble the three genomes that were originally used in the study, confirming that KBase can be used to reliably perform metagenomic analysis workflows that previously would have entailed transferring data between multiple systems.
4. Future Work
KBase is in active development, and we plan to continue extending and improving its capabilities. Our longer-term plan is to realize the promise of KBase as a knowledgebase. We will enhance the social aspects of the platform, enabling users to find others with similar or complementary interests and to form KBase groups to simplify collaboration in the system. We will also add data-discovery features such as the ability to suggest datasets and Narratives that are likely to be of interest to a user. We will link KBase to high-performance-computing resources at DOE to enable analyses to be performed across all system data and facilitate users in performing more complex, larger studies. Ultimately, the aim is to enable KBase to propose new hypotheses by finding connections between wide-ranging results stored in the system.

5. Conclusions
KBase provides a range of tools and datasets in an integrated environment that can be used to create, run and share reproducible systems biology analysis workflows operating on large or heterogeneous data. These capabilities can be explored interactively via the exemplar Narratives mentioned in this report. An increasing number of researchers are using KBase in their work; publications that cite KBase in their methods can be found at https://www.kbase.us/research. As more users apply the system to address their scientific questions, and share their resulting Narratives, KBase will have a continually growing body of experiments, results and scientific use cases that can be adapted and extended by other researchers.
The system is open source and open access, and is designed to be extensible by the community. KBase’s Software Development Kit (SDK) enables external developers to incorporate third-party open source tools as KBase apps. Such third-party apps are beginning to appear in the system, and the pace of development is increasing.
KBase is actively engaging the external community to help us improve our tools and workflows in all areas of computational system biology. We welcome your feedback and contributions, which could take the form of public Narratives, third-party apps, publications citing use of KBase, or even talks or workshops to train your colleagues to use KBase. With your help, KBase will become an ever more useful community resource.
Summary Report Authors:
Nomi Harris
KBase PIs:
Adam Arkin, Robert Cottingham, Chris Henry
Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nature Biotechnology. 2018;36: 566. doi: 10.1038/nbt.4163
- Arkin, A.P. et al. The DOE Systems Biology Knowledgebase (KBase). Preprint at bioRxiv, http://biorxiv.org/content/early/2016/12/22/096354 (2016). DOI: http://dx.doi.org/10.1101/096354.
- Pérez F, Granger BE. IPython: A System for Interactive Scientific Computing, Computing in Science and Engineering, vol. 9, no. 3, pp. 21-29, May/June 2007, doi:10.1109/MCSE.2007.53. URL: http://ipython.org
- Wilkinson, M.D. et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 3, 160018 (2016).
- Georganas, E., et al., HipMer: An Extreme-Scale De Novo Genome Assembler. Proceedings of Sc15: The International Conference for High Performance Computing, Networking, Storage and Analysis, 2015.
- https://jgi.doe.gov/data-and-tools/bbtools/
- Li, D., et al., MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, 2015. 31(10): p. 1674-6.
- Nurk, S., et al., Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol, 2013. 20(10): p. 714-37.
- Peng, Y., et al., IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics, 2012. 28(11): p. 1420-8.
- Gurevich, A., et al., QUAST: quality assessment tool for genome assemblies. Bioinformatics, 2013. 29(8): p. 1072-5.
- Brettin, T., et al., RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep, 2015. 5: p. 8365.
- Overbeek, R., et al., The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Research, 2005. 33(17): p. 5691-5702.
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014 Jul 15;30(14):2068-9. https://www.ncbi.nlm.nih.gov/pubmed/24642063
- Henry, C.S., et al., High-throughput generation, optimization, and analysis of genome-scale metabolic models. Nature Biotechnology, 2010. Nbt.1672: p. 1-6.
- Latendresse, M., Efficiently gap-filling reaction networks. BMC Bioinformatics, 2014. 15: p. 225.
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter, L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562 578. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3334321/
- Pertea M, Kim D, Pertea G, Leek JT and Salzberg SL (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown. Nature Protocols 11, 1650–1667. http://www.nature.com/nprot/journal/v11/n9/full/nprot.2016.095.html
- Price, M.N., P.S. Dehal, and A.P. Arkin, FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One, 2010. 5(3): p. e9490.
- Camacho, C., et al., BLAST+: architecture and applications. BMC Bioinformatics, 2009. 10: 421.
- Eddy, S.R., Accelerated Profile HMM Searches. PLoS Comput Biol, 2011. 7(10): p. e1002195.
- Wu, Y.W., et al., MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome, 2014. 2: p. 26.
- Parks, D.H., et al., CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res, 2015. 25(7): p. 1043-55.
- Wu, X., et al., Draft Genome Sequences of Two Janthinobacterium lividum Strains, Isolated from Pristine Groundwater Collected from the Oak Ridge Field Research Center. Genome Announce., 2017. 5 (26)
- Li, L., C.J. Stoeckert, Jr., and D.S. Roos, OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res, 2003. 13(9): p. 2178-89.
- Edirisinghe JN, Weisenhorn P, Conrad N, Xia F, Overbeek R, Stevens RL, Henry CS. Modeling central metabolism and energy biosynthesis across microbial life. BMC Genomics. 2016;17. doi:10.1186/s12864-016-2887-8
- Magnusdottir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol 35, 81-89 (2017).
- Weston, D.J., et al., Sphagnum physiology in the context of changing climate: emergent influences of genomics, modelling and host–microbiome interactions on understanding ecosystem function. Plant, cell & environment, 2015. 38(9): 1737-51.
- Marshall, C.W., et al., Metabolic Reconstruction and Modeling Microbial Electrosynthesis. Sci Rep, 2017. 7(1): p. 8391.
